
بیماری رتینوبلاستوما چیست؟
زمان مورد نیاز برای مطالعه : 14 دقیقه
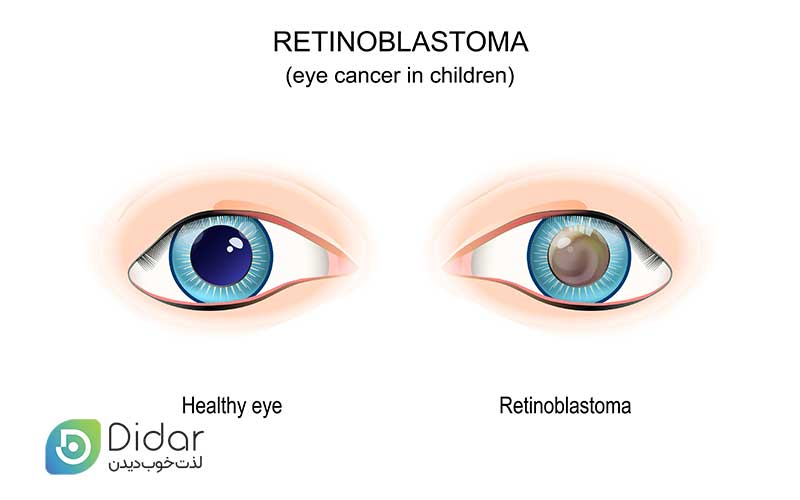
رتینوبلاستوما یک سرطان چشم در دوران کودکی است. این بیماری نادر است و میزان بقای کلی بسیار بالایی دارد. زمانی اتفاق میافتد که سلولهای شبکیه پشت چشم بهطور غیرقابل کنترلی تکثیر میشوند. تشخیص زودهنگام علائم، مانند مردمک چشم سفید یا کم رنگ (لکوکوریا)، همانطور که به ویژه در انعکاس چشم در عکس ها دیده می شود، احتمال یک نتیجه خوب را بهبود می بخشد.
رتینوبلاستوما چیست؟
رتینوبلاستوما نوعی سرطان چشم است که در شبکیه چشم شما، لایه حساس به نور از سلول های پشت چشم شما شروع می شود. این شایع ترین سرطان چشم در دوران کودکی است.
رتینوبلاستوما می تواند در یک یا هر دو چشم ایجاد شود. از هر 4 مورد 1 مورد هر دو چشم را درگیر می کند. کارشناسان گمان می کنند که این اتفاق به دلیل نقص در سلول های جوان و در حال رشد شبکیه رخ می دهد. در 4 مورد از 5 مورد، تشخیص قبل از 3 سالگی رخ می دهد. در موارد نادر، بزرگسالان نیز می توانند پس از وقفه در رشد اولیه تومور دچار این بیماری شوند.
شبکیه از بافت عصبی تشکیل شده است که نور را هنگام ورود از جلوی چشم حس می کند. نور باعث می شود شبکیه سیگنال هایی را به مغز ارسال کند. مغز سیگنال ها را به صورت تصویر تفسیر می کند.
رتینوبلاستوما اغلب در کودکان خردسال اتفاق می افتد. معمولاً قبل از 2 سالگی تشخیص داده می شود. اغلب یک چشم را درگیر می کند. گاهی اوقات در هر دو چشم اتفاق می افتد. چندین روش درمانی برای رتینوبلاستوما وجود دارد. برای اکثر کودکان، درمان نیازی به برداشتن چشم برای رهایی از سرطان ندارد. چشم انداز کودکان مبتلا به رتینوبلاستوما بسیار خوب است.
انواع رتینوبلاستوما
سه نوع رتینوبلاستوما وجود دارد:
- یک طرفه: این به معنای “یک طرفه” است، بنابراین فقط یک چشم را تحت تأثیر قرار می دهد.
- دو طرفه: این به معنای “دو طرفه” است، بنابراین هر دو چشم را تحت تاثیر قرار می دهد.
- سه جانبه: این بدان معناست که شما در سه نقطه سرطان دارید. هر چشم یکی از آن مکان ها را منعکس می کند. سومین مکان در غده صنوبری در داخل مغز شما قرار دارد. (سرطانی که آن غده را تحت تأثیر قرار می دهد پینئوبلاستوم نامیده می شود.)
حدود 60 درصد موارد رتینوبلاستوما یک طرفه است. موارد دو طرفه و سه جانبه 40 درصد دیگر را تشکیل می دهند.
رتینوبلاستوما چقدر شایع است؟
رتینوبلاستوما نادر است. حدود 3.3 مورد در هر 1 میلیون نفر زیر 20 سال وجود دارد. در این گروه سنی، سالانه کمی بیش از 300 مورد جدید در ایالات متحده و کمی کمتر از 9000 مورد جدید در سراسر جهان وجود دارد.
علائم و نشانه های بیماری رتینوبلاستوما
از آنجایی که این بیماری معمولاً قبل از 3 سالگی تشخیص داده می شود، کودکان اغلب نمی توانند علائم خود یا آنچه را که تجربه می کنند را توصیف کنند. در عوض، علائم تغییرات قابل مشاهده در ظاهر چشم یا تفاوت در نحوه رفتار کودک شما است.
لوکوکوریا:
اولین و شایع ترین علامت رتینوبلاستوما این است که مردمک چشم شما سفید (لکوکوریا) یا رنگ پریده در تنظیمات خاص به نظر می رسد، به ویژه در عکس هایی که در مکان های کم نور گرفته شده است و همچنین از فلاش برای روشنایی استفاده می شود. ممکن است در یک یا هر دو چشم اتفاق بیفتد.
سایر علائم رتینوبلاستوما:
در حالی که رتینوبلاستوما معمولاً در کودکان قبل از صحبت کردن اتفاق می افتد، علائم و نشانه های دیگری نیز وجود دارد که می تواند نشان دهنده رشد آن باشد، از جمله:
- چشم هایی که در پیگیری حرکت مشکل دارند یا اصلا آن را دنبال نمی کنند.
- ناهماهنگی چشم (استرابیسم)
- درد (ممکن است باعث شود که کودک شما بیشتر از حد معمول گریه کند یا مزاحم باشد، یا ممکن است در خواب یا تغذیه مشکل داشته باشد)
- بزرگ شدن چشم (بوفتالموس)
- برآمدگی چشم (پروپتوز)
- خون در محفظه جلویی چشم شما (هایفما)
- عفونت، تورم یا التهاب چشم یا بافت اطراف (سلولیت اربیتال)
چه چیزی باعث رتینوبلاستوما می شود؟

رتینوبلاستوما نوعی سرطان است که در آن سلولهای شبکیه نادرست عمل کرده و شروع به تکثیر غیرقابل کنترل میکنند. همانطور که انجام می دهند، می توانند تومور ایجاد کنند و به بافت های اطراف آسیب برسانند. اگر آنها بدون کنترل به رشد خود ادامه دهند، در نهایت، آن سلول های ناکارآمد فراتر از تومور اصلی گسترش می یابند (متاستاز می دهند) و سرطان در سایر نقاط بدن شما ظاهر می شود.
نقصی که باعث می شود سلول های سالم به رتینوبلاستوما تبدیل شوند در DNA شما شروع می شود.
رتینوبلاستوما چگونه ایجاد می شود؟
چشم ها قبل از تولد شروع به رشد می کنند. در مراحل اولیه رشد، چشم ها دارای سلول هایی به نام رتینوبلاست هستند که با تکثیر سلول های جدیدی ایجاد می کنند که شبکیه را پر می کنند. در یک نقطه خاص، این سلول ها از تکثیر باز می ایستند و به سلول های بالغ شبکیه تبدیل می شوند. به ندرت اتفاقی در این روند رخ می دهد. به جای بلوغ، برخی از رتینوبلاست ها به رشد خارج از کنترل ادامه می دهند و سرطانی به نام رتینوبلاستوما را تشکیل می دهند.
زنجیره رویدادهای درون سلولی که منجر به رتینوبلاستوما می شود پیچیده است، اما تقریباً همیشه با تغییر (جهش) در ژن RB1 شروع می شود. ژن طبیعی RB1 به جلوگیری از رشد خارج از کنترل سلول ها کمک می کند، اما تغییر در ژن مانع از عملکرد آن می شود. بسته به زمان و مکان تغییر در ژن RB1، می تواند منجر به 2 نوع مختلف رتینوبلاستوما شود.
نقش DNA در به وجود آمدن بیماری رتینوبلاستوما
سلول های شما از DNA مانند یک کتاب آشپزی استفاده می کنند. شما DNA را از هر دو والدین بیولوژیکی خود به ارث می برید، بنابراین مانند این است که هر کدام بخشی از کتاب آشپزی خود را به شما می دهند، بنابراین می توانید یک کتاب آشپزی کامل برای خود بسازید.
اما گاهی اوقات ممکن است در DNA خود دچار خطا شوید. سلولهای شما فقط میدانند چگونه دستور العملهای موجود در کتاب را دقیقاً دنبال کنند، بنابراین یک خطای DNA به این معنی است که سلولهای شما فقط میدانند چگونه دستور پخت را اشتباه درست کنند. که می تواند باعث رشد و تکثیر غیرقابل کنترل آن سلول ها شود.
متخصصان توصیه می کنند که برای کودکان مبتلا به رتینوبلاستوما، صرف نظر از ارثی بودن یا نبودن آن، آزمایش ژنتیک و مشاوره انجام شود. آنها همچنین آزمایش برای خواهر و برادرهای بیولوژیکی و سایر اعضای خانواده را توصیه می کنند.
جهشی که باعث رتینوبلاستوما می شود، RB1، یک ژن سرکوبگر تومور را تحت تاثیر قرار می دهد. ژن های سرکوبگر تومور مسئول رشد طبیعی بافت ها (شبکیه شبکیه) هستند. آنها مانند ترمز عمل می کنند و تولید مثل و فرآیندهای رشد سلولی را کنترل می کنند. جهش در RB1 به این معنی است که سلول های شبکیه شما می توانند بدون کنترل به تومور تبدیل شوند. رتینوبلاستوما همچنین می تواند با “حذف” کروموزوم 13p، که محل ژن RB1 است، رخ دهد.
در موارد نادر، افراد ممکن است یک رشد خوش خیم شبکیه به نام رتینوم داشته باشند. اینها مانند پیش سازهای رتینوبلاستوما هستند، اما به دلایلی رشدشان متوقف می شود. در اواخر زندگی، رتینوم ممکن است دوباره شروع به رشد کند و به رتینوبلاستوما تبدیل شود.
خطاهای DNA که باعث ایجاد رتینوبلاستوما می شوند دو راه اصلی وجود دارد:
پراکنده:
این زمانی است که یک خطایی رخ می دهد در حالی که سلول های شما DNA یک والدین را کپی می کنند. مانند این است که هنگام کپی کردن دستی یک دستور، اشتباه تایپی را به طور تصادفی معرفی کنید. دستوری که از آن کپی کردید درست بود، اما خطای جدید به این معنی است که دستور پخت شما اکنون نادرست است. موارد پراکنده رتینوبلاستوما فقط یک چشم را تحت تاثیر قرار می دهد.
ارثی:
این زمانی است که یک یا هر دو والدین بیولوژیکی دارای DNA با خطا هستند. سلول های شما DNA خود را به درستی کپی می کنند، اما این شامل اشتباه تایپی از قبل موجود نیز می شود. رتینوبلاستوما دارای توارث اتوزومال غالب است. این به این معنی است که اگر یکی از والدین این ژن را داشته باشد، تقریباً 50٪ احتمال دارد و اگر هر دو والدین آن را داشته باشند، 75٪ احتمال دارد. اما والدین بیولوژیکی ممکن است رتینوبلاستوما نداشته باشند، حتی اگر فرزندشان فرم ارثی داشته باشد. این به این دلیل است که برخی از افراد ناقل هستند، به این معنی که آنها جهش را دارند بدون اینکه باعث ایجاد این بیماری در آنها شود.
به ارث بردن یک جهش ژنی همچنین تعیین می کند که فرزند شما چه شکلی از رتینوبلاستوما دارد. موارد ارثی معمولا دو طرفه و کمتر یک طرفه هستند. خواهر و برادر کودک مبتلا به رتینوبلاستوما نیز شانس بیشتری برای ابتلا به آن دارد. وقتی هر دو والدین تحت تأثیر رتینوبلاستوما قرار نگیرند، خواهر و برادرهای بیولوژیکی کودک مبتلا نیز 4 تا 7 درصد خطر ابتلا به رتینوبلاستوما را دارند.
عوارض رتینوبلاستوما
رتینوبلاستوما می تواند به بافت های اطراف آسیب برساند. می تواند باعث نابینایی جزئی یا کامل در چشم(های) آسیب دیده شود.
از آنجا که این یک نوع سرطان است، خطر گسترش رتینوبلاستوما (متاستاز) به سایر قسمت های بدن شما نیز وجود دارد. زمانی که این اتفاق بیفتد، حتی خطرناکتر میشود، بنابراین جلوگیری از گسترش آن بخش کلیدی درمان است. یکی از راه های خطرناک انتشار آن از طریق عصب بینایی به مغز شماست، جایی که به یک تومور سرطانی جدید مغز تبدیل می شود.
جهش های ژنتیکی که می تواند باعث ایجاد رتینوبلاستوما شود، خطر ابتلا به انواع دیگر سرطان را نیز افزایش می دهد. خطر تجمعی 1% در سال برای ابتلا به سرطان دیگر وجود دارد (مثلاً حدود 20% در 20 سالگی). محتمل ترین سرطان ها عبارتند از:
- سارکوم: اینها بر استخوان ها و بافت های همبند تأثیر می گذارد.
- ملانوم: اینها بر پوست، چشم ها و غشاهای مخاطی شما مانند غشاهای داخل دهان و بینی تأثیر می گذارند.
- سرطانهای ریه: به دلیل اتصالات پیچیده رگهای خونی در ریهها، سرطانها در اینجا به راحتی میتوانند در هر جای دیگری از بدن پخش شوند.
رتینوبلاستوما چگونه تشخیص داده می شود؟
والدین ممکن است اولین کسانی باشند که لکوکوریا را می بینند و به متخصص اطفال کودک خود می گویند، او نیز می تواند آن را جستجو کند. یک متخصص اطفال میتواند لکوکوری را در طول معاینه تأیید کند، یا ممکن است در طول معاینات فیزیکی معمولی که رشد کودک را نظارت میکند، لکوکوری را ببیند.
هنگامی که یک متخصص اطفال لکوکوریا را مشاهده کرد، گام بعدی معمولاً ارجاع فوری به چشم پزشک یا سایر متخصصان مراقبت از چشم است. یک متخصص چشم سعی می کند با نگاه کردن به داخل چشم مستقیماً رتینوبلاستوما را ببیند. برای کودکان خردسال، این ممکن است شامل استفاده از قطره های دارویی برای گشاد کردن مردمک چشم یا انجام معاینه چشم تحت بیهوشی باشد.
اسکن های تصویربرداری محتمل است، زیرا این اسکن ها می توانند به دنبال تومورهای سخت تر در چشم دیگر یا تومورهای مربوطه در مغز (مانند پینئوبلاستوما) باشند. اسکن های تصویری که ممکن است از آنها استفاده کنند عبارتند از:
سونوگرافی:
این اسکن می تواند تجمع کلسیم را نشان دهد که در رتینوبلاستوما شایع است.
توموگرافی کامپیوتری (CT) اسکن:
رتینوبلاستوما اغلب شامل تجمع کلسیم است که در سی تی اسکن نیز قابل مشاهده است.
اسکن تصویربرداری رزونانس مغناطیسی (MRI):
این بهترین اسکن برای تصاویر دقیق از بافت ها و ساختارهای مختلف داخل بدن شما است. بیشتر طول می کشد و گران تر است، بنابراین اغلب اولین آزمایش نیست. با این حال، این یک راه حیاتی برای مشاهده میزان گسترش تومور یا تشخیص تومور در نقاط دیگر چشم یا مغز دیگر است.
اسکن توموگرافی گسیل پوزیترون (PET):
این آزمایش ممکن است در مراحل اولیه تشخیص و درمان یا مدت طولانی بعد از آن انجام شود. این به ویژه برای تشخیص اینکه آیا تومور به مکان های دیگر متاستاز داده است یا اینکه تومورهای دیگر در جای دیگری ایجاد شده اند مفید است.
روش های درمان رتینوبلاستوما
راه های متعددی برای درمان رتینوبلاستوما وجود دارد. اغلب، درمان شامل ترکیبی از روش ها است. آنها می توانند به طور همزمان یا متوالی اتفاق بیفتند. روش های درمانی عبارتند از (اما محدود به موارد زیر نیست):
شیمی درمانی:
در این روش از داروهایی استفاده می شود که به طور مستقیم به نقاط ضعف سلول های سرطانی حمله می کنند. این می تواند به جلوگیری از جراحی که ممکن است شامل از دست دادن بینایی باشد کمک کند. همچنین می تواند باعث کوچک شدن تومورها به اندازه ای شود که سایر درمان ها می توانند سلول های سرطانی باقی مانده را از بین ببرند.
این می تواند موضعی باشد، به این معنی که از طریق تزریق هدفمند یا تزریق از طریق شریان (داخل شریان) اتفاق می افتد. همچنین می تواند سیستمیک باشد، به این معنی که از طریق انفوزیون استاندارد داخل وریدی (IV) اتفاق می افتد. روش تجویز بسته به مورد خاص و نیاز شما متفاوت است.
پرتودرمانی:
این شامل استفاده از انرژی با فرکانس بالا برای از بین بردن سلول های تومور است. این می تواند از خارج از بدن شما با درمان هایی مانند پرتودرمانی خارجی (EBRT) یا در داخل بدن شما با روش هایی مانند براکی تراپی اتفاق بیفتد. با این حال، درمان معمولاً به دلیل خطرات طولانی مدت عوارض از این امر جلوگیری می کند.
درمان های کانونی (کرایوتراپی، گرما درمانی، لیزر درمانی و غیره):
درمان های کانونی به این دلیل نام خود را گرفته اند که بر تخریب مستقیم سلول های تومور تمرکز می کنند. این می تواند شامل گرما درمانی یا لیزر درمانی باشد که از انرژی گرمای شدید برای از بین بردن سلول ها استفاده می کند یا سرما درمانی که از سرمای شدید برای از بین بردن سلول ها به جای گرما استفاده می کند.
جراحی:
جراحی به احتمال زیاد زمانی انجام می شود که خطر گسترش رتینوبلاستوما وجود داشته باشد. این معمولاً شامل جراحی مانند انوکلئاسیون است که به معنای برداشتن چشم است. این از گسترش بیشتر سرطان جلوگیری می کند. در زمانی که این جراحی ضروری است، سرطان احتمالاً به اندازه کافی به چشم آسیب دیده شما آسیب رسانده است که باعث از دست دادن بینایی می شود.
سایر درمان ها و درمان ها:
این روش ها می توانند بسیار متفاوت باشند و معمولاً به عوارض جانبی سایر درمان ها کمک می کنند. یکی از نمونههای درمان مانند این، داروهایی است که میتواند به تهوع و استفراغ کمک کند، که در شیمیدرمانی رایج است. از آنجایی که درمانهای حمایتی زیادی مانند این وجود دارد، پزشک متخصص چشم شما بهترین فردی است که به شما میگوید چه چیزی را برای مورد خاص و نیازهای شما توصیه میکند.
آیا می توان از رتینوبلاستوما پیشگیری کرد؟
رتینوبلاستوما به دلیل جهش های ژنتیکی اتفاق می افتد، بنابراین هیچ راهی برای پیشگیری کامل از آن وجود ندارد. اگر سابقه خانوادگی رتینوبلاستوما دارید یا میدانید که حامل جهش ژنتیکی هستید که باعث آن میشود، مشاوره ژنتیک میتواند به شما در درک خطر انتقال آن به یک کودک بیولوژیکی کمک کند.
در صورت داشتن این شرایط، چه انتظاری باید داشت؟
چشم انداز رتینوبلاستوما به شدت به مدت زمان تشخیص و درمان آن بستگی دارد، اما به طور کلی احتمال آن بسیار خوب است. میزان بقای کلی برای رتینوبلاستوما کودکان 95٪ است. شانس یک نتیجه خوب – از جمله اجتناب از از دست دادن بینایی – با تشخیص قبل از 2 سالگی بهترین است.
افرادی که از رتینوبلاستوما جان سالم به در می برند برای سرطان های جدید به مراقبت مادام العمر نیاز دارند. این معمولاً شامل اسکن سالانه یا آزمایشهای دیگری است که میتواند تومورهای جدید را تشخیص دهد. متخصص چشم شما می تواند به شما بگوید که چه اقدامات نظارتی را برای پرونده شما توصیه می کند.
چه زمانی باید به پزشک برای رتینوبلاستوما مراجعه کرد؟
اگر متوجه علائم رتینوبلاستوما یا تغییر در چشم یا بینایی فرزندتان شدید، با پزشک خود تماس بگیرید. اگر شما یا شریک زندگیتان سابقه خانوادگی رتینوبلاستوما دارید یا می دانید که جهش ژن RB1 دارید، با پزشک خود صحبت کنید. ممکن است بخواهید قبل از بچه دار شدن، مشاوره ژنتیک را در نظر بگیرید.
در صورت داشتن سابقه خانوادگی رتینوبلاستوما چه چیزی باید بدانم؟
اگر سابقه خانوادگی رتینوبلاستوما دارید، حتماً معاینات منظم چشم را برای فرزندتان و سایر اعضای خانواده برنامه ریزی کنید. ژن RB1 که باعث رتینوبلاستوما می شود، همچنین می تواند باعث ایجاد رتینوسیتوم، یک تومور خوش خیم (غیر سرطانی) چشم شود. رتینوسیتوم می تواند در افراد در هر سنی ایجاد شود.
جمع بندی
رتینوبلاستوما شایع ترین شکل سرطان دوران کودکی است، اما خوشبختانه نادر است. و زمانی که این اتفاق بیفتد، شانس کلی برای یک نتیجه خوب بالا است، به خصوص با تشخیص و درمان زودهنگام.
والدین و سایر مراقبان در تشخیص و تشخیص زودهنگام رتینوبلاستوما نقش کلیدی دارند. مطمئن شوید که فرزندتان به طور منظم معاینه می شود و اگر متوجه انعکاس مردمک چشم سفید یا کم رنگ غیرمعمول در عکس های کودکتان شدید، سریع با پزشک اطفال او صحبت کنید. انجام این کار می تواند به فرزند شما کمک کند در صورت نیاز مراقبت شود.

